Заболевания, связанные с нарушением обмена соединительной ткани
Выделяют три основных компонента соединительной ткани:
■ клеточные элементы;
■ коллагеновые, эластические и ретикулярные волокна;
■ аморфное основное вещество.
Генетические дефекты синтеза и распада различных структурных компонентов соединительной ткани приводят к развитию наследственных
заболеваний. Среди заболеваний, при которых поражаются коллагеновые и эластические волокна, наиболее часто наблюдают болезнь Марфана. Группа заболеваний, в основе которых лежит нарушение метаболизма аморфного вещества, в основном кислых гликозаминогликанов, получили название мукополисахаридозы.
Болезнь Марфана — наследственное заболевание, характеризующееся системным поражением соединительной ткани. В развитии болезни важное значение имеет поражение коллагена и эластина, выражающееся в нарушении внутри- и межмолекулярных связей в этих структурах. Первичный генетический дефект, обусловливающий болезнь Марфана, связан с повреждением гена фибриллина-1 (#154700, 15q15-q21.3, ген FBN1 [*134797], . В норме этот ген кодирует синтез соединительнотканного протеина — фибриллина-1 (структурный гликопротеинный компонент эластина). Для больных типичны высокий рост, длинные (паукообразные) пальцы, бедренные и паховые грыжи, гипоплазия мышц и подкожной клетчатки, плоскостопие, пороки сердца, аневризма аорты.
Мукополисахаридозы. Важнейшая составная часть соединительной ткани — протеогликаны (крупные макромолекулы, состоящие из волокнистого центрального белка с ковалентно присоединёнными к нему гликоза-миногликанами). Количество и соотношение различных протеогликанов зависит от типа соединительной ткани. Они образуются в фибробластах. В лизосомах этих же клеток протеогликаны после эндоцитоза разрушаются. Разрушение гликозаминогликанов начинается с терминального моносахарида под действием специфических лизосомальных гликозидаз. Если какие-либо лизосомальные ферменты отсутствуют (генетический дефект их синтеза) или их активность снижена, в соединительной ткани различных органов начинается накопление неразрушенных или частично разрушенных протеогликанов. Поэтому мукополисахаридозы относятся к болезням накопления.
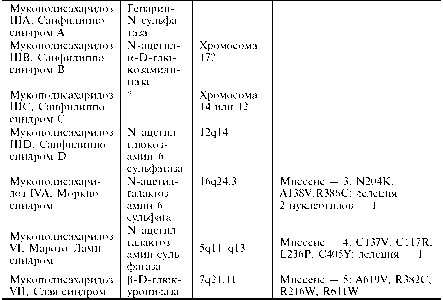
В настоящее время выделяют 9 основных типов мукополисахаридозов, характеризующихся недостаточностью различных ферментов и определёнными особенностями клинической картины. Перечень основных типов мукополисахаридозов, дефектные ферменты, хромосомная локализация генов, типы и количество мутаций приведены в табл..
Таблица Молекулярно-генетические основы мукополисахаридозов
Таблица Молекулярно-генетические основы мукополисахаридозов

- Подагра лечение народными средствами в домашних условиях
- Сбор мокроты, мочи, кала для лабораторного исследования
- Лечение авитаминоза народными средствами и методами
- Лечение токсикоза народными средствами и методами
- Лечение склероза народными средствами и методами
- Лечение женских болезней народными средствами