Заболевания, связанные с нарушением обмена гема и порфиринов
Талассемии — гетерогенная группа генетических нарушений, связанных с нарушением синтеза нормальных полипептидных цепей Hb. Эти аномалии приводят к развитию гипохромных микроцитарных анемий различной тяжести. Снижение синтеза а-глобиновых цепей вызывает а-талассемию, Р-цепей — р-талассемию. У человека два идентичных гена а-глобина на каждой хромосоме 16 и по одному гену Р-глобина на хромосоме 11.
Большинство случаев а-талассемии обусловлены делецией генов, контролирующих синтез а-глобиновых цепей. Тяжесть а-талассемии связана с количеством делеций этих генов. Делеция одного гена клинически не проявляется. При делеции двух генов возможна лёгкая анемия со снижением MCV. Результаты электрофореза Hb нормальны. Для установления диагноза используют методы ДНК-диагностики. При потере 3 генов развивается анемия различной степени выраженности с содержанием Hb 70-90 г/л. При электрофорезе выявляют HbH Делеция всех 4 генов приводит к несовместимой с жизнью водянке плода.
Р-Талассемия (*141900, 11p15.5, более 90% всех талассемий) развивается в результате экспрессии аномальных генов р-глобиновой цепи. Малая Р-талассемия возникает у лиц, гетерозиготных по патологическому гену. У большинства из них клинические проявления отсутствуют, однако выявляют гипохромию эритроцитов и снижение MCV. Диагноз подтверждаётся путём проведения электрофореза Hb (повышенное содержание HbA2 и/или HbF). Большая р-талассемия (анемия Кули) развивается у лиц, гомозиготных по патологическому гену. Уже на первом году жизни развивается тяжёлая гипохромная микроцитарная анемия. Лечение включает регулярные переливания крови в сочетании с введением железосвязывающих препаратов или пересадку красного костного мозга.
Порфирии — группа гетерогенных, преимущественно наследственных заболеваний, в основе которых лежат нарушения биосинтеза гема и накопление в организме порфиринов и/или их предшественников.
Синтез гема происходит в 8 этапов, для каждого из которых необходим специфический фермент. Ключевую роль играет фермент первого этапа — синтетаза аминолевулиновой кислоты, регуляция её активности лимитирует скорость синтеза гема. Под влиянием индуцирующих факторов активность этого фермента может повышаться в 5-6 раз, а при накоплении конечного продукта (гема) она снижается.
Форма порфирии определяется недостаточностью одного из 8 специфических ферментов цепи синтеза гема. Ферментативный блок на любом уровне данной цепи приводит к снижению количества гема и вызывает повышение активности синтетазы аминолевулиновой кислоты. В результате происходит накопление продуктов синтеза перед заблокированным участком цепи.
В зависимости от того, где происходит повышенное образование и накопление порфиринов и их предшественников, порфирии разделяются на печёночные и эритропоэтические. В большинстве случаев ферментативный дефект выражен во всех тканях, поэтому правильнее говорить лишь о преимущественном вовлечении в процесс либо печени, либо костного мозга. Референтные величины концентрации порфиринов приведены в табл..
Таблица Референтные величины концентрации порфиринов в крови [Henry J.D., 1996]
Таблица Референтные величины концентрации порфиринов в крови [Henry J.D., 1996]

По клиническому течению порфирии разделяют на острые (ОПП, наследственная копропорфирия и др.) и хронические (врождённая порфи-рия, кожная печёночная порфирия и др.).
Наиболее распространённый вариант — ОПП (частота — приблизительно 1:30 000), связанная с недостаточностью гидроксиметилбилан синтета-зы, порфобилиноген дезаминазы или уропорфироген синтетазы (*176000, 11q23.3, дефекты генов HMBS, PBGD, UPS).
У 70-90% носителей патологического гена ни разу в жизни не возникает каких-либо клинических проявлений. В остальных случаях болезнь проявляется приступами острых болей в животе, поражениями периферической (полиневропатия) и центральной (судороги, эпилептиформные припадки, бред, галлюцинации) нервной системы, провоцируемых принятием ряда ЛС и гормональных препаратов, а также различными стрессами, с возможным летальным исходом (летальность приблизительно 60%). ОПП относится к группе острых печёночных порфирий.
Предположительный диагноз острой порфирии может быть поставлен на основании появления окрашенной мочи во время приступа (от слегка розовой до красно-бурой). Розовый цвет мочи обусловлен повышенным содержанием в ней порфиринов, а красно-бурый — присутствием про-топорфирина, продукта деградации порфобилиногена. Для подтверждения диагноза проводят комплекс биохимических и генетических исследований.
На первом этапе исследуют мочу на присутствие в ней избытка порфо-билиногена — качественный скрининговый тест с реактивом Эрлиха, или
58-аминолевулиновой кислоты (5-АЛК). Порфобилиноген, реагируя с реактивом Эрлиха, образует в кислом растворе окрашенный продукт розово-красного цвета. Этот тест почти всегда положителен при острых приступах порфирии и лишь в редких случаях бывает ложноположительным. Отрицательный результат теста не позволяет исключить диагноз острой порфи-рии. Это обусловлено целым рядом причин: в моче могут присутствовать вещества-ингибиторы, обусловливающие ложноотрицательный результат; повышение концентрации порфобилиногена может быть незначительным (ниже предела чувствительности метода); экскреция порфобилиногена может быстро снижаться и нормализоваться в течение нескольких дней после острого приступа. В связи с этим все положительные и некоторые отрицательные (при наличии соответствующей клинической картины заболевания) результаты теста должны быть подтверждены количественным определением порфобилиногена в моче. Учитывая, что в некоторых случаях при ОПП вначале резко повышается содержание 5-АЛК, необходимо при наличии клинических симптомов и отрицательной пробы на порфобили-ноген провести исследование на 5-АЛК.
В норме концентрация порфобилиногена в моче меньше 2 мг/л. При получении нормальных результатов количественного определения пор-фобилиногена в моче порфирию как причину острых симптомов можно в большинстве случаев отвергнуть. У пациентов с увеличенной концентрацией ПГБ в моче устанавливают диагноз «острая порфирия» и в дальнейшем выполняют исследования по дифференциальной диагностике ОПП и других форм острой порфирии. Для этих целей используется определение общих порфиринов в кале.
В норме концентрация общих порфиринов в кале меньше 200 ммоль/кг сухого кала. Нормальная концентрация общих порфиринов в кале подтверждает диагноз ОПП. При вариегатной порфирии и врождённой прото-порфирии эта концентрация увеличивается во много раз.
Таким образом, диагноз ОПП в период острого течения заболевания можно установить на основании повышенной концентрации порфобили-ногена в моче и нормального содержания общих порфиринов в кале. Вне обострения и в бессимптомных случаях повышенное содержание порфо-билиногена в моче выявляют только у 30% больных ОПП. В таких случаях необходимо провести исследование активности порфобилиноген дезами-назы в эритроцитах.
Порфобилиноген дезаминаза — цитоплазматический фермент, катализирующий конденсацию четырёх молекул порфобилиногена с образованием линейного тетрапиррола. Фермент существует в двух изоформах, одна из которых специфична для эритроцитов, а другая содержится в клетках практически всех тканей. В норме активность порфобилиноген дезамина-зы в эритроцитах — 5,8-11,7 нмоль/с/л [Тиц Н., 1997]. Приблизительно у 90% больных ОПП активность фермента в эритроцитах снижена в 2 раза. Приблизительно у 5% пациентов активность порфобилиноген дезаминазы может быть в пределах нормальных величин из-за перекрывания уровней активности фермента в норме и при ОПП. В таких случаях точный диагноз может быть поставлен только с использованием молекулярно-генети-ческих методов. Информативность различных методов диагностики ОПП
в зависимости от периода заболевания представлена в табл.-10-7, а на Рис. приведён алгоритм диагностики заболевания. Ген порфобилино-ген дезаминазы локализован на хромосоме 11 (11q23-11qter). Для выявления мутаций исследуют ДНК лимфоцитов больных методом ПЦР, секве-нирования или ПДРФ-анализа.
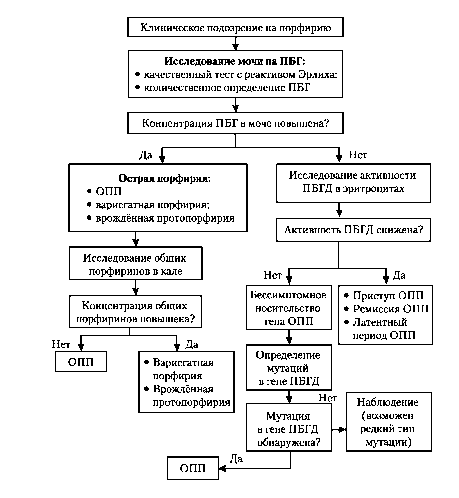
Рис. Алгоритм обследования пациентов с подозрением на порфирию
Рис. Алгоритм обследования пациентов с подозрением на порфирию
Таблица Информативность различных методов диагностики ОПП в зависимости от периода заболевания
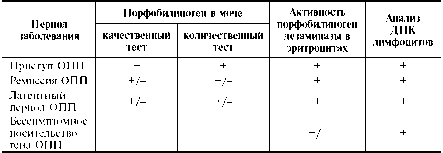
Таблица Типичные биохимические изменения, ассоциированные с нарушениями метаболизма порфирина [Henry J.D., 1996]
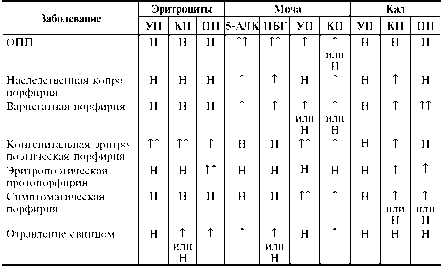
Примечания: УП — уропорфирин; КП — копропорфирин; ПП — протопорфи-рин; 5-АЛК — 58-аминолевулиновая кислота; ПБГ — порфобилиноген; Т — повышение; ТТ — значительное повышение; Н — норма.
Примечания: УП — уропорфирин; КП — копропорфирин; ПП — протопорфи-рин; 5-АЛК — 58-аминолевулиновая кислота; ПБГ — порфобилиноген; Т — повышение; ТТ — значительное повышение; Н — норма.